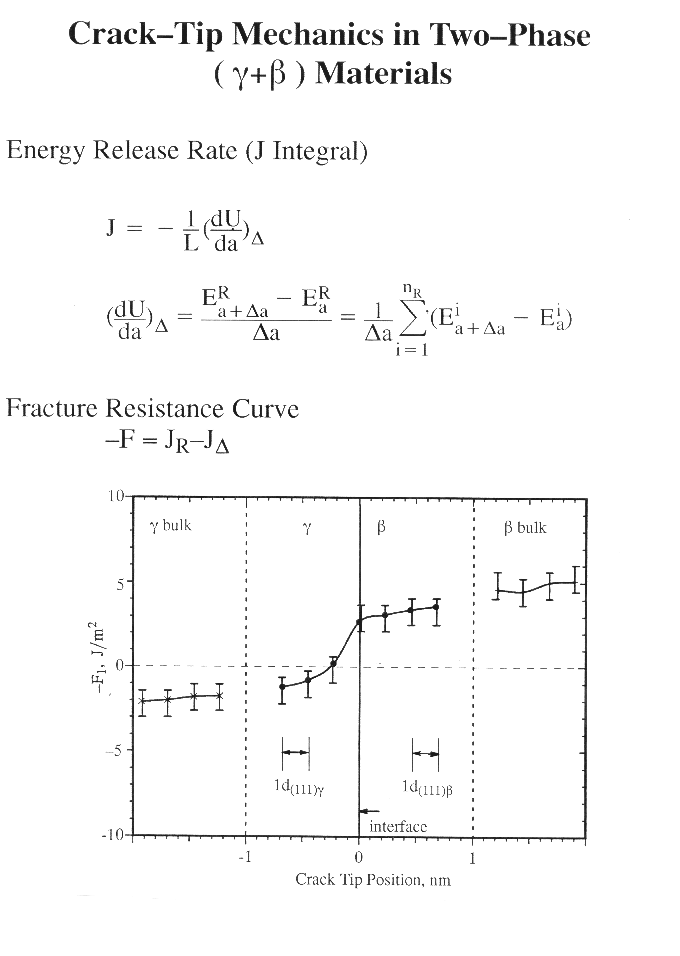
It is well-established that the materials evolution at the crack-tip is not directly coupled to the applied load but rather is screened from it by various additional sources of internal stress such as the dislocations, second phase precipitates, microcracks, etc. The fracture process is an event occuring at the atomistic scale and hence atomistic simulations of fracture are expected to provide a useful supplement to the more more conventional elastic/plastic continuum analyses of fracture.
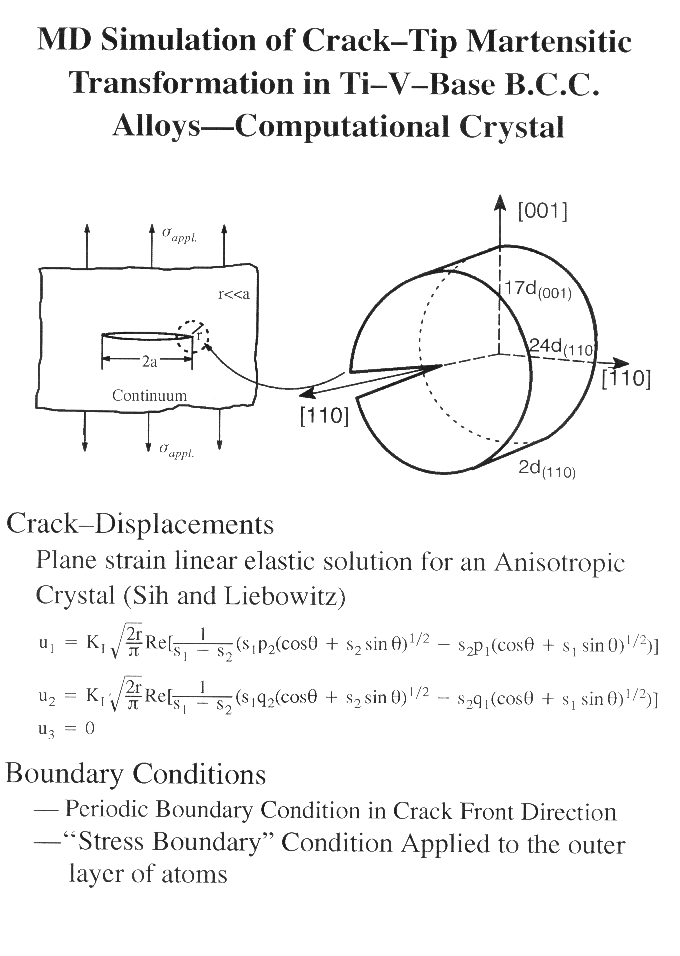
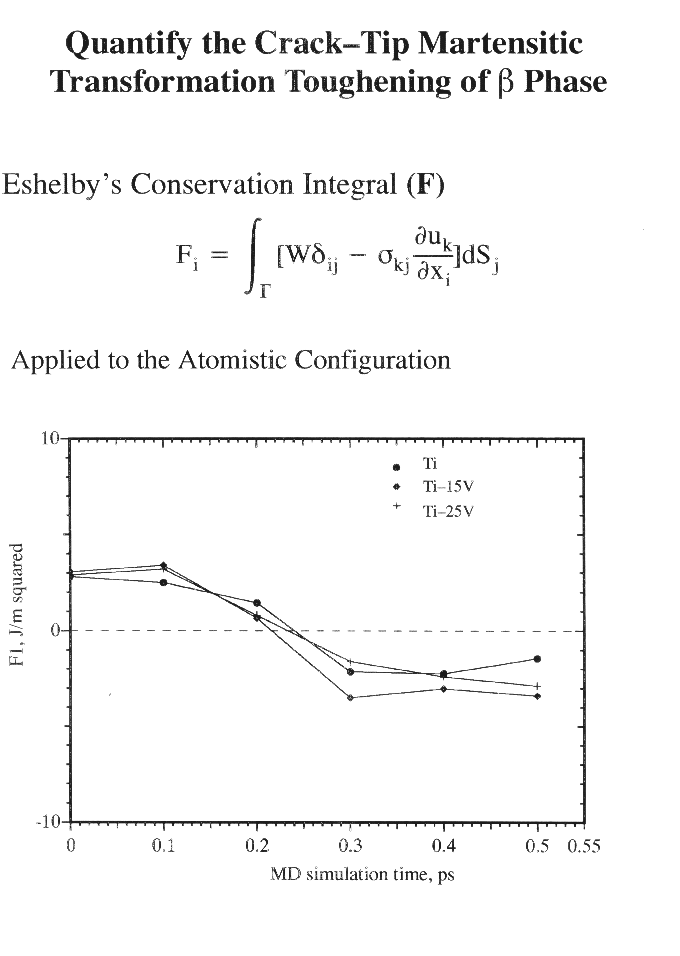
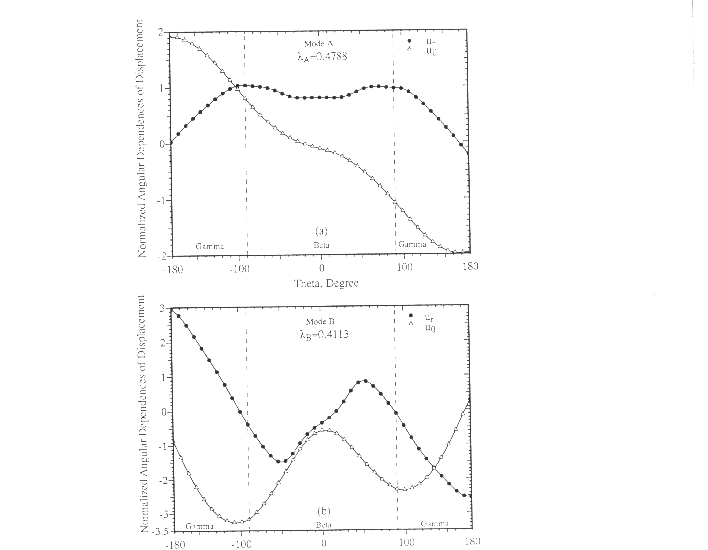
The present analysis deals a crack of length 2a subject to a tensile load normal to the crack plane applied ata an infinite materials (Mode I). An atomistic cylindrical (or rectangular) crystal of a radius r (r is much smaller than a) centered at the crack tip is selected as the computational crystal in the MD simulations. The effect of the surrounding bulk is accounted for by applying the "fixed stress boundary conditions" on the outer layer of atoms in the computational crystal. A periodic boundary condition is employed in the crack front direction, and hence the analysis is nominally a plane strain problem. Molecular Dynamics (MD) simulations are carried out to simulate the crack propagation process. Fracture Mechanics and FEM methods are combined to generate the initial computational crystal and to quantify the toughness of an atomistic crystal under transforming.
1. P. Dang and M. Grujicic, "The Effect of Crack-tip Materials Evolution on Fracture Toughness-An Atomistic Simulation Study of the Ti-V alloy System", Acta Metallurgica et Materialia, 75-87, 45 (1997).
2. M. Grujicic and P. Dang, "A Molecular Dynamics Study of Transformation Toughening in the Gamma TiAl/Beta Ti-V System", Materials Science and Engineering A, 109-125, 219 (1996).
3. P. Dang and M. Grujicic, "Atomic Simulation of Crack-tip Transformation in Ti-V beta Alloy", Scripta Metallurgica et Materialia, 59-64, 35 (1996).
4. M. Grujicic and P. Dang, "Molecular Dynamics/Embedded Atom Method Simulation of Crack-tip Transformation Toughening in Fe-Ni Austenite", Materials Science and Engineering A, 173-182, 199 (1995).
![]()
{kind=link}
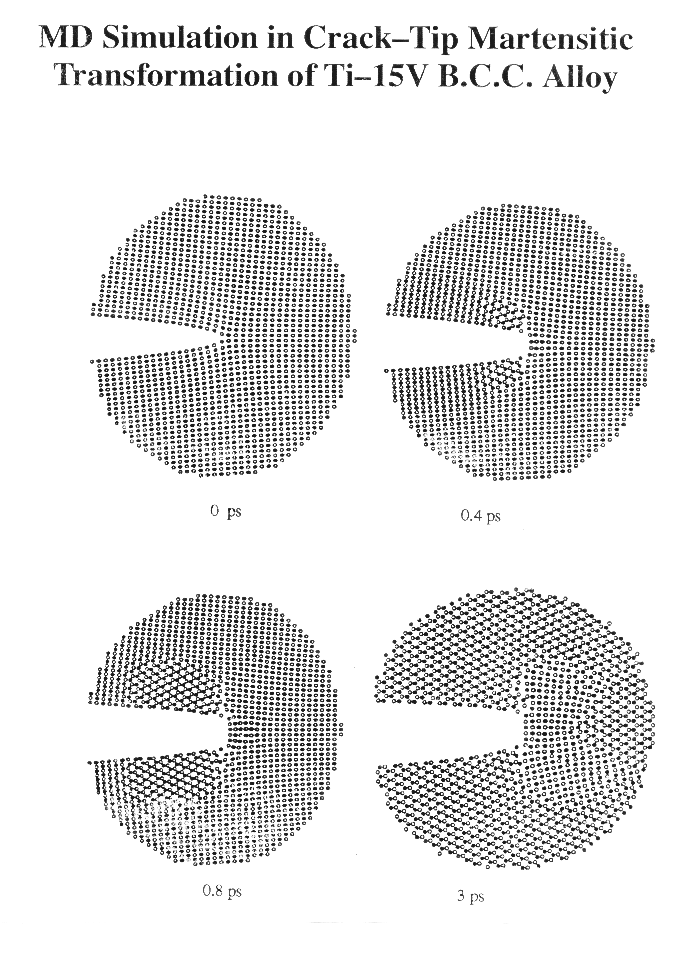{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}